 |
| Home | Research | Publications | Prospective Students | Teaching | Contact | Physics | Resources | Fun |
| Research Home
Current Projects Other Projects |
Protein Misfolding and Aggregation-Related Disease
Aggregation-related diseases include Alzheimer's, Amyotrophic Lateral Sclerosis (ALS), Huntington's disease, and the Prion diseases such as Creutzfeld-Jacob disease (CJD). They all have no cure at present. These diseases in humans range from rare in the case of CJD (with an incidence rate of about 1 in a million humans) to common in the case of Alzheimer's (about 10% of those over 65 and about 50% over 85). However they all share a common general mechanism involving neurodegeneration: healthy functional protein misfolds and aggregates, during which process oligomeric species recruit and induce healthy protein to misfold via a template-directed process. Intriguingly, essentially all the neurodegerative disease-related proteins are involved in metal binding and metal-mediated catalysis. For example Superoxide dismutase (SOD1), the homodimeric protein whose misfolding is responsible for the symptoms of ALS, normally functions as an anti-oxidant by converting negatively-charged superoxide radicals to hydrogen peroxide and molecular oxygen through a two-step catalytic process involving the successive reduction and oxidation of a copper ion embedded in the core of the protein. This rate for this reaction is among the fastest known for all enzymes making the reaction essentially diffusion-limited.
Using antibodies designed to mimic the action of misfolded prion (PrPSc), our experimental collaborators have been able to induce healthy prion (PrPc) to misfold, and have dissected which regions are involved in misfolding [1]. These regions turn our to have thermodynamic signatures that are computationally tractable using statistical mechanics analysis along with a proper energy function [2].
One mystery that emerged from the experiments was a kind of "action at a distance" problem wherein an antibody that bound to a region on the N-terminal unstructured tail of PrPc induced the misfolding (as probed by exposure of previously buried epitopes to subsequence diagnostic antibody binding) of a region on the first C-terminal alpha-helix.
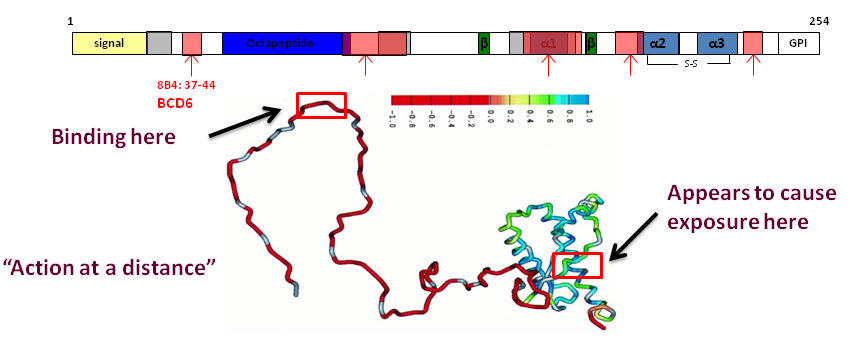
We hypothesized in [1] that the disordered N-terminal tail is not in a random-coil state, but instead must exist in a more condensed phase around the structured domain of the protein, similar to a "molten shell".
This condensed halo of polymer is not sufficiently structured to yield any significant NMR constraints, but must contribute to the stability of the C-terminal structured domain through the collective effect of transient interactions again imperceptible to conventional NMR analysis. The C-terminal domain can be thought of as an "avidity-enhanced structure".
Misfolding of superoxide dismutase (SOD1)
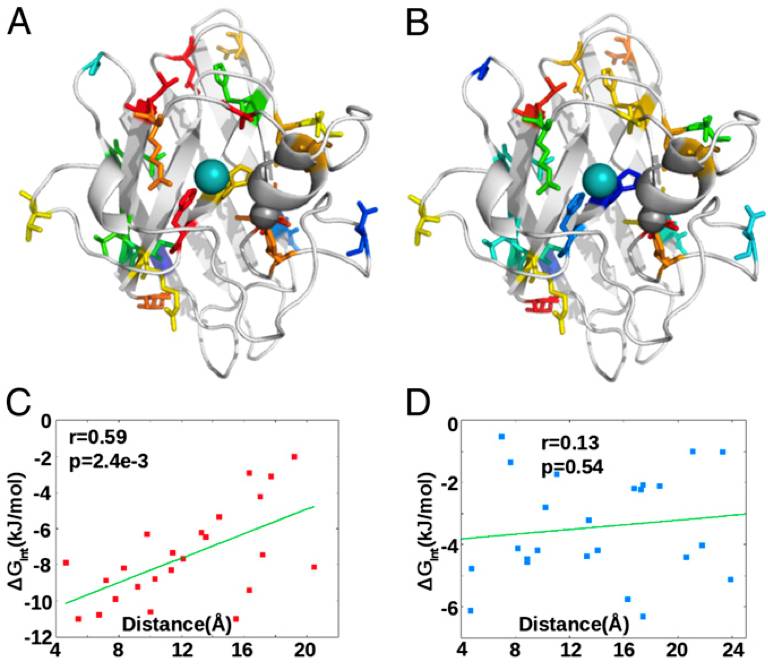
More recently, we have investigated the allosteric interaction-free energies between residues and metals in the protein SOD1. Related to this, we have simulated a series of atomic force microscopy experiments with variable tether positions to quantify mechanical rigidity “fingerprints” for SOD1 variants. Mechanical fingerprinting studies of a series of C-terminally truncated mutants, along with an analysis of equilibrium dynamic fluctuations while varying native constraints, potential energy change upon mutation, frustratometer analysis, and analysis of the coupling between local frustration and metal binding interactions for a glycine scan of 90 residues, together revealed that the apo protein is internally frustrated, that these internal stresses are partially relieved by mutation but at the expense of metal-binding affinity, and that the frustration of a residue is directly related to its role in binding metals. This evidence pointed to apo SOD1 as a strained intermediate with “self-allostery” for high metal-binding affinity.
Thus, the prerequisites for the function of SOD1 as an antioxidant compete with apo state thermo-mechanical stability, increasing the susceptibility of the protein to misfold in the apo state.That is, the very function of this protein puts it in peril for misfolding!
In collaboration with our experimental colleagues in the Cashman group, we have been investigating the intermolecular induction of protein misfolding-- the central phenomenon of protein misfolding disease.
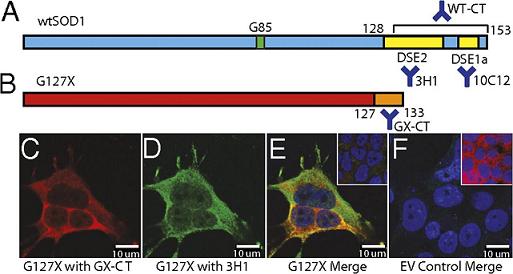
The above figure shows schematic sequence overviews of (A) wtSOD1 and (B) G127X SOD1, a truncation mutant with a non-native C-terminal sequence of 6 amino acids (orange); an antibody (GX-CT) can be designed that binds to this sequence. Such an antibody will probe for the presence of G127X SOD1, but not wtSOD1. Residue G85 is mutated in some FALS cases and is shown in green.
Misfolded SOD1 displays several cryptic epitopes that are normally hidden, called disease-specific epitopes or DSEs. Our experimental colleagues have found some of these, whose locations are noted in yellow, along with the corresponding antibodies (3H1 and 10C12).
Panel (C) shows an immunofluorescence (IF) image of G127X-transfected HEK cells probed with GX-CT polyclonal IgG, specific for the nonnative C terminus of G127X SOD1. Panel (D) shows that in the same cells, full-length SOD1 has misfolded, because it was probed with a mAb specific to full-length SOD1. Panel (E) shows the merge of C and D with the nuclear-specific stain DAPI.
Thus, cells positive for full-length misfolded SOD1 immunoreactivity (3H1) also coexpress G127X, so it appears that the expression of G127X mutant SOD1 induces wtSOD1 to misfold!
There is essentially no "base-level" misfolding of SOD1 in these cells, i.e. empty vector transfected cells stained with GX-CT, 3H1, and DAPI don't show any misfolded full-length SOD1 (Panel F), while they do express natively-folded SOD1, as seen by staining with DAPI and pan-SOD1 antibody (Panel F inset).
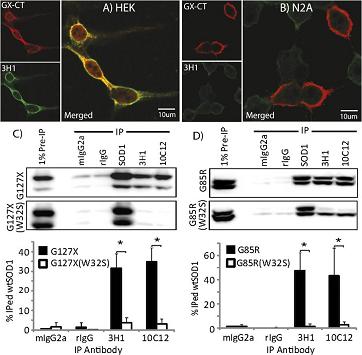
Panels (A) and (B) in the above figure show IF images of HEK cells (A) expressing human SOD1 and mouse N2a (B) cells expressing mouse SOD1, that have been transfected with (human) G127X gene, and probed with GX-CT and 3H1 (the full-length misfolding-specific mAb).
The HEK cells expressing G127X display 3H1 show immunoreactvity for misfolded wtSOD1, but the mouse cells do not.
It's worth noting from the sequence alignment between mouse and human SOD1:

that the mAbs specific for SOD1 misfolding would have bound to mouse SOD1 had it misfolded.
Panel (C) above shows an immunoprecipitation (IP) study of lysates from HEK cells transfected with either the mutant G127X (top), or with G127X/W32S , i.e. with the tryptophan at position 32 mutated to a serine (bottom).
The key lanes to look at are the ones where full-length misfolded SOD1 specific mAbs are the bead bound Abs (two lanes on the far right). They pulled down misfolded WT SOD1, as expected from the study above. Moreover, they also pulled down G127X (bottom band)-- so G127X must have been bound (either dimerically or oligomerically) to wtSOD1.
But-- the single mutation W32S ablates the ability of mutant SOD1 to misfold wtSOD1! There are no bands for the full-length misfolding-specific SOD1 Ab pulldown.
This effect is quantified in the bottom of the panel. The story is essentially recapitulated with the mutant G85R.
Thus there is a "species-barrier" for the misfolding of SOD1: human misfolded SOD1 will not convert mouse SOD1, and moreover the essence of the "species-barrier" can be localized to one amino acid, tryptophan 32!
The effect works the other way as well: G127X will not convert full length SOD1 to misfold if full length SOD1 has the mutation W32S.
Certainly something is special about W32-- the above studies point to a critical homotypic interaction as essential for intermolecular transduction of misfolding. We are now employing computational studies to investigate the structural and energetic consequences of these inter-molecular interactions.
References:
[1] Li L, Guest W, Huang A, Plotkin SS, Cashman N, "Immunological mimicry of PrPc-PrPSc interactions: Antibody-induced PrP misfolding" Protein Engineering, Design and Selection 22(8):523-529 (2009).
[2] Guest W, Cashman N, Plotkin SS, "Electrostatics in the Stability and Misfolding of the Prion Protein: Salt Bridges, Self-Energy, and Solvation" Biochem. Cell Biol 88, 371–381 (2010)
[3] Guest W, Plotkin SS, Cashman NR, "Toward A Mechanism of Prion Misfolding and Structural Models of PrPSc: Current Knowledge and Future Directions", J Toxicol Env Health Part A, 74:154–160 (2011)
[4] Grad LI, Guest W, Yanai A, Pokrishevsky E, O’Neill MA, Gibbs E, Semenchenko V, Yousefi M, Wishart DS, Plotkin SS, Cashman NR, "Intermolecular transmission of superoxide dismutase 1 misfolding in living cells" Proc. Natl. Acad. Sci, 108, 16398–16403 (2011)
[5] Das A, Plotkin SS "SOD1 Exhibits Allosteric Frustration to Facilitate Metal Binding Affinity" Proc. Natl. Acad. Sci. USA 110, 3871 (2013)
[6] Das A, Plotkin SS "Mechanical Probes of SOD1 Predict Systematic Trends in Metal and Dimer Affinity of ALS-Associated Mutants" J. Mol. Biol. 425, 850–874 (2013)
[7] Das A, Sin BK, Mohazab AR, and Plotkin SS "Unfolded protein ensembles, folding trajectories, and refolding rate prediction" J. Chem. Phys. 139, 121925 (2013) http://jcp.aip.org/resource/1/jcpsa6/v139/i12/p121925_s1
[8] Grad LI, et al "Intercellular propagated misfolding of wild-type Cu/Zn superoxide dismutase occurs via exosome-dependent and -independent mechanisms" Proc. Natl. Acad. Sci. USA 111, 3620-3625 (2014) http://www.pnas.org/content/111/9/3620.full.pdf+html?with-ds=yes
Home | Research | Publications | Prospective Students | Teaching | Contact | Physics | Resources || Fun |